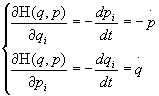 i=1,
,3N i=1,
,3N
i=1,
,3N i=1,
,3N
(Umberto Lucia)*
La Meccanica Statistica Classica (una descrizione rigorosa dovrebbe fare riferimento alle leggi della Meccanica Statistica Quantistica. La Meccanica Statistica Classica è un caso particolare della prima, valido quando la descrizione quantistica tende a quella classica: h® 0 , con h costante di Planck, 6.67 10-34 Js . Nell'approccio termodinamico la teoria cinetica classica permette di descrivere con notevoli risultati i fenomeni sperimentalmente osservati o osservabili, quindi per quanto riguarda la descrizione termodinamica è sufficiente ricorrere alla teoria classica della Meccanica Statistica) è un settore della Fisica sviluppato col fine di analizzare le proprietà della materia quando essa si trova in equilibrio termodinamico.
Il fine di questa disciplina è di derivare tutte le proprietà macroscopiche del sistema in esame dalle leggi della dinamica microscopica del sistema stesso: si opera in questo modo una descrizione macroscopica del sistema ponendone le basi fisiche su un'analisi microscopica. La Meccanica Statistica non fornisce una descrizione di come un sistema raggiunge l'equilibrio, né determina le condizioni fisiche per cui un sistema permanga in equilibrio, ma descrive lo stato di equilibrio quando esso sia già stato raggiunto. In questo contesto risulta interessante definire per mezzo della dinamica microscopica le grandezze macroscopiche che forniscono informazioni riguardo allo stato di equilibrio e le sue condizioni di realizzabilità, come per esempio l'entropia. Infatti, dopo aver definito una grandezza macroscopica dalla quale si possano derivare le condizioni di equilibrio e di stabilità del sistema, si possono applicare a questa funzione le leggi della termodinamica connesse con lo stato di equilibrio e le condizioni di stabilità stesse.
Ogni stato del sistema, costituito da N particelle, è univocamente e completamente definito da un insieme di 3N coordinate canoniche {q1, ..., qN} e di 3N impulsi canonici {p1, ..., pN}: queste 6N coordinate costituiscono la base dello spazio delle fasi (q,p) 6N-dimensionale, o spazio G, del sistema. La dinamica del sistema è determinata dalla hamiltoniana H(q,p) dalla quale si possono ottenere le equazioni canoniche del moto
i=1,
,3N i=1,
,3N
Ogni punto nello spazio delle fasi rappresenta uno stato fisico e questa
relazione è biunivoca. Inoltre il luogo geometrico dei punti che
soddisfa l'equazione
H(q,p) = E
è un'ipersuperficie nello spazio delle fasi di definita energia E. In termini di operatori matematici l'energia E è un autovalore dell'operatore di evoluzione eiH, nel caso classico, oppure dell'operatore hamiltoniano, in quello quantistico, e lo stato che essa caratterizza è un autostato. Lo stato del sistema evolve secondo le equazioni canoniche del moto e descrive una curva nello spazio delle fasi che si trova sempre sulla stessa ipersuperficie di energia in quanto il sistema è conservativo.
Per un sistema macroscopico l'interesse termodinamico è focalizzato
solo su alcune specifiche proprietà macroscopiche del sistema stesso
come per esempio il volume definito V e l'energia E compresa
in un determinato intervallo [E,E+DE].
Queste proprietà macroscopiche sono soddisfatte da un notevole numero
di possibili stati microscopici del sistema, i microstati, pertanto si
introduce una funzione che esprime per il volume elementare d3Nq
d3Np dello spazio delle fasi la probabilità
di realizzazione dello specifico stato macroscopico, la densità
di probabilità r(q,p;t).
Essendo interessati alla situazione di equilibrio occorre limitare la propria
attenzione alla descrizione la cui funzione di densità non dipende
esplicitamente dal tempo t, ma solo dalle variabili canoniche (q,p)
per mezzo dell'hamiltoniana H(q,p)
cioè:
r(q,p;t) = r(q,p) = r(H(q,p)) (1)
da cui segue, dal teorema di Liouville, che la densità di probabilità
r(q,p;t) è
una grandezza conservata, quindi costante nel tempo. Infatti secondo il
teorema di Liouville si ha che:
![]() (2)
(2)
ma le parentesi di Poisson per r(H(q,p)) sono nulle, pertanto anche ¶r/¶t = 0 .
Inoltre nella teoria statistica si introduce il postulato di uguaglianza della probabilità a priori secondo il quale "quando un sistema macroscopico è in equilibrio termodinamico, il suo stato microscopico deve essere con uguale probabilità uno qualsiasi degli stati che soddisfano le condizioni macroscopiche del sistema stesso". Conseguenza di questo postulato è che il sistema considerato sia un elemento di un insieme detto insieme microcanonico con la funzione densità di probabilità definita come:

In questo contesto se si considera un'osservabile, definendo come tale
una qualsiasi grandezza termodinamica misurabile direttamente o indirettamente,
O(q,p) ,
il suo valore misurato deve corrispondere al suo valore medio calcolato
nell'insieme microcanonico. Il suo valore più probabile è
quello realizzato dal maggior numero di microstati corrispondenti al determinato
macrostato considerato, mentre il valore medio risulta:
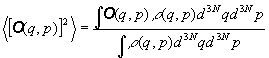 (3)
(3)
Inoltre il valore medio e quello più probabile coincidono se
la fluttuazione quadratica media è molto piccola, cioé se:
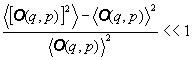 (4)
(4)
Se quest'ultima condizione non è verificata, allora non è possibile determinare univocamente il valore osservato della grandezza fisica considerata e, quando ci si trova in questa condizione la Meccanica Statistica non è più applicabile perchè non sono più verificate le ipotesi su cui è fondata. In tutti i casi fisici di interesse per la termodinamica la fluttuazione quadratica media è dell'ordine dell'inverso del numero di particelle N che costituiscono il sistema termodinamico, ma essendo molto grande tale numero, allora la fluttuazione quadratica media è molto prossima allo 0, quindi la condizione di equivalenza tra valore più probabile e valore medio nell'insieme microcanonico è verificata.
Come conseguenza questo permette di sviluppare una teoria statistica della Termodinamica e di definire attraverso una funzione di probabilità la funzione di stato entropia. Per fare ciò occorre innanzitutto definire a livello microscopico l'entropia e verificare che essa corrisponde alla funzione di stato termodinamico entropia.
Si consideri il volume G(E) occupato
nello spazio delle fasi dall'insieme microcanonico:
![]() (5)
(5)
e quello, S(E) , racchiuso dalla superficie
di energia E:
![]() (6)
(6)
allora, come conseguenza del postulato di uguaglianza della probabilità
a priori, si ottiene la relazione:
G(E) = S(E + DE) - S(E) (7)
e se DE << E si può
sviluppare S(E + D
E)? in serie di McLaurin al primo ordine, ottenendo:
![]() (8)
(8)
dove si è indicato con w il coefficiente dello sviluppo del primo ordine.
Con queste relazioni si introduce, in Meccanica Statistica Classica,
la definizione di entropia come:
S(E,V) = kB ln(G(E)) (9)
dove kB è la costante di Boltzmann. Questa definizione statistica di entropia deve essere equivalente a quella classica questo significa che deve godere delle stesse proprietà analitiche, deve contenere le stesse informazioni globali e locali sul sistema e deve permettere di giungere agli stessi risultati fisici, cioè ai principi della termodinamica.
Innanzi tutto l'entropia (3) è una grandezza estensiva, cioè se un sistema è composto di due sottosistemi, le cui entropie sono S1 e S2 , la sua entropia totale S risulta la somma di quelle dei sottosistemi stessi, S = S1 + S2 (altrimenti detto, si definisce estensiva ogni grandezza il cui valore è funzione delle massa del sistema). Per dimostrare che anche la (9) soddisfa questa proprietà si considera un sistema composto da due sottosistemi che hanno rispettivamente N1 e N2 particelle ed i volumi V1 e V2 . L'energia di interazione molecolare tra i due sottosistemi è trascurabile se confrontata con l'energia totale di ogni sottosistema.
L'hamiltoniana totale del sistema composto è la somma delle hamiltoniane
dei due sottosistemi, cioè:
H(q,p) = H1((q,p)1) + H2((q,p)2) (10)
Si considera, quindi, per prima cosa il caso in cui i due sottosistemi
siano tra loro isolati: essi hanno energia rispettivamente compresa il
primo nell'intervallo [E1,E1+D
E1] ed il secondo in [E2,E2+D
E2]. Le entropie dei sottosistemi, in base alla (9),
sono:
S(E1,V1) = kB ln(G1(E1)) (11)1
S(E2,V2) = kB ln(G2(E2)) (11)2
Ora si prende in esame l'insieme microcanonico del sistema composto e si pone l'energia nell'intervallo [E1,E1+2DE1] e si sceglie DE << E in modo che valga l'approssimazione effettuata precedentemente per ottenere l'espressione di G(E) in funzione di DE . Questo insieme microcanonico contiene tutte le coppie del sistema composto per cui:
1. le N1 particelle le cui posizioni nello spazio delle fasi sono (q,p)1 sono contenute nel volume V1 ;
2. le N2 particelle le cui posizioni nello spazio delle fasi sono (q,p)2 sono contenute nel volume V2 ;
3. le energie E1 ed E2 dei sottosistemi
presentano valori che soddisfano la condizione:
E1 + E2 Î [E1+E2,E1+E2+2DE] (12)
Il volume dello spazio delle fasi che corrisponde alle condizioni di
equilibrio termico con l'energia totale che si trova nell'intervallo [E1+E2,E1+E2+2DE]
è:
G(E1)G(E2) (13)
Se i due sistemi sono energeticamente isolati rispetto all'esterno,
posti a contatto essi raggiungono l'equilibrio oltre che con l'esterno
anche tra loro: con l'accoppiamento non si verifica alcuno scambio di energia,
cioè:
dG(E1) = dG(E2) = 0 (14)
e quindi in particolare:
dlnG(E1) = dlnG(E2) = 0 (15)
pertanto si ha che:
d(lnG(E1)G(E2)) = 0 (16)
Se vi fosse uno scambio di calore, però, si avrebbe una variazione
in G(E) e quindi anche in lnG(E)
e questa variazione sarebbe proporzionale al calore scambiato. Se una quantità
di calore (-DQ1), come definito
nella Termodinamica dell'equilibrio, si trasferisce dal sistema 1 al sistema
2 si avrebbe che:
DQ1 = ?DQ2 = 0 (17)
e, quindi:
![]() (18)
(18)
da cui si ottiene:
Dln[G1(E1)G2(E2)] = (b2 - b1)DQ (19)
In questo caso la condizione di equilibrio risulta:
b2 £ b1 (20)
Se si ipotizza che il trasferimento di calore avvenga dal sistema 2
al sistema 1, allora con considerazioni equivalenti si giunge alla condizione
di equilibrio:
b2 ³ b1 (21)
Affinché queste due condizioni di equilibrio siano contemporaneamente
verificate nella situazione fisica considerata deve essere soddisfatta
la relazione:
b2 = b1 (22)
Poiché la condizione di equilibrio termodinamico è legata alla uguaglianza del valore delle temperature - equilibrio termico - allora si rileva un legame tra i coefficienti b e la temperatura stessa. Questo prova la proprietà estensiva dell'entropia, perché:
Dln[G1(E1?)G2(E2?)] = (b2-b1)DQ = D[lnG1(E1)-lnG2(E2?)] (24)
Inoltre questa dimostrazione prova anche il fatto che i sottosistemi
si trovano rispettivamente negli stati di energia +E1
, e +E2
. Questi sono i valori di energia che rendono massima la funzione G1(E1)G2(E2)
con la condizione E = E1 + E2,
cioè analiticamente:
![]() (25)
(25)
da cui si ricava la condizione:
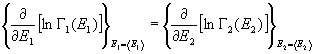 (26)
(26)
cioè:
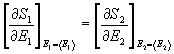 (27)
(27)
Da questi risultati con l'aggiunta di temperatura assoluta, si deduce che la temperatura di un sistema isolato è il parametro che governa l'equilibrio tra i sottosistemi, cioè si trova la condizione di equilibrio termico.
Inoltre l'entropia, definita dalla relazione statistica (9):
S(E,V) = kB lnG(E) = kB lnw(E) = kB lnS(E) (28)
è una funzione sempre crescente in quanto S(E) è una funzione non decrescente, pertanto tale risulta anche la funzione entropia.
Si è così verificato che l'entropia nella definizione
statistica coincide con l'entropia così come espressa nella sua
definizione classica.
Bibliografia
* ITIS "Alessandro Volta"
Spalto Marengo, 42
15100 Alessandria
Società Italiana di Storia delle Matematiche
Dipartimento di Matematica, Università di Torino
Via Carlo Alberto 10, 10100 Torino, Italy
- - - - -
Umberto Lucia è nato ad Alessandria nel
1966. Laureato in Fisica a Torino nel 1991, si è successivamente
perfezionato in Fisica della Materia, ed ha acquisito il dottorato di ricerca
in Energetica (VIII ciclo, Firenze e Roma, 1996). E' docente di ruolo di
Fisica presso l'ITIS "Volta" di Alessandria e svolge ricerche sia in Storia
della Matematica presso il Dipartimento di Matematica di Torino sia in
Metodi matematici della Fisica-matematica e della Chimica Teorica. E' stato
Tecnologo a tempo determinato presso il Nucleo Applicativo (afferente alla
Direzione Generale) dell'Istituto Nazionale per la Fisica della Materia,
dove si è occupato di Ricerca Applicata. Ha insegnato "Metodologie
fisiche per i beni culturali" per il corso di Storia del restauro presso
l'Università degli Studi di Bologna, sede di Ravenna (Corso di laurea:
Conservazione dei Beni Culturali). E' socio fondatore della SISM - Società
Italiana di Storia delle Matematiche. Organizza e gestisce alcune attività
della formazione post-secondaria nella Provincia di Alessandria: corsi
post-diploma e IFTS; progetti di trasferimento tecnologico dalla ricerca
all'industria; ha contribuito allo studio della rete di trasferimento tecnologico
dell'INFM ed ha proposto le attività di ricerca sulle Metodologie
fisiche per i beni culturali, "da Parnaso a Giano"; ha collaborato con
l'ENEA alla realizzazione del Simposio "Meccanica statistica e termodinamica
computazionale: dai fondamenti alle applicazioni in campo ingegneristico,
ambientale e diagnostico" nell'ambito del IV Congresso Nazionale della
SIMAI - Società Italiana di Matematica Applicata ed Industriale
- realizzato a Giardini Naxos (Messina), 1-5 giugno 1998. Oltre a quelli
relativi ai suoi specifici campi di competenza, ha interessi in: Fondamenti
della Fisica e della Matematica; Didattica della Fisica e della Matematica;
Cibernetica e Teoria dei Sistemi. Collabora con i settimanali locali Il
Piccolo alessandrino e La Voce alessandrina, per la diffusione
della cultura scientifica. E' membro del direttivo del Centro Studi
"Francesco Faà di Bruno" di Alessandria, ente morale legalmente
riconosciuto, dove si occupa di problemi sociali e culturali.
E-mail: fismat@libero.it